蛋白|一天之内,两大AI预测蛋白结构算法开源,分别登上Nature、Science
机器之心报道
机器之心编辑部
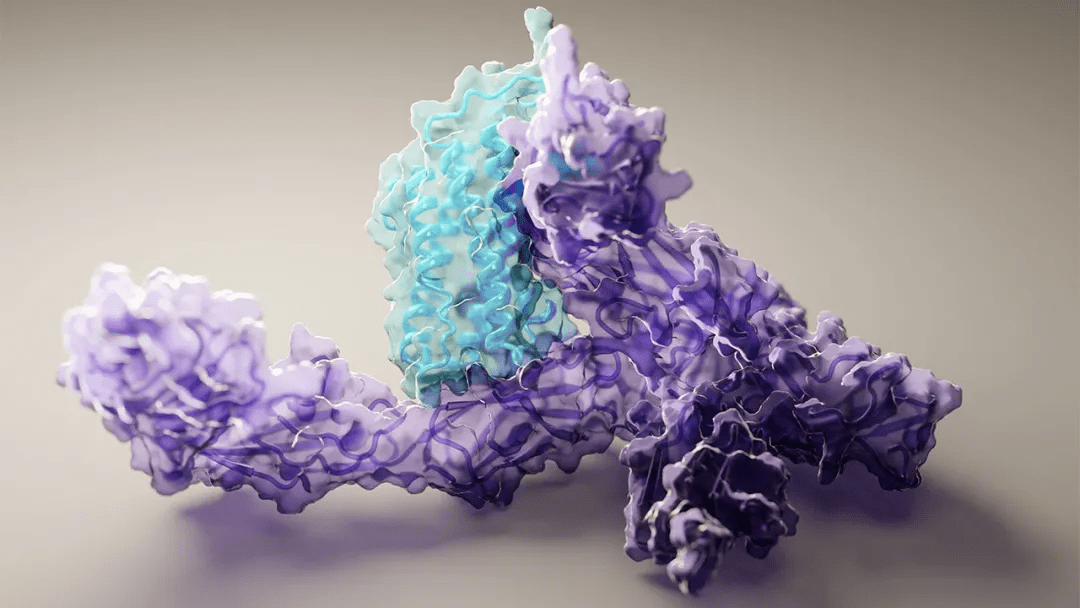
这是科学激动人心的新一步 。使用 DNA 序列预测蛋白质形状的 AphaFold2 , 终于开源了 。
众所周知 , 蛋白质是生命活动的基本组件 , 它们可以单独存在 , 也会协同工作 。 为了发挥作用 , 这些长链氨基酸扭曲、折叠并交织成复杂的形状 , 这些形状可能很难 , 甚至根本不可能破译 。
科学家们一直在梦想通过基因序列简单地预测蛋白质形状——如果能够成功 , 这将开启一个洞察生命运作机理的新世界 。 然而近五十年来 , 人们的进展缓慢 。
7 月 15 日 , 《自然》杂志一篇论文被接收的消息引发了人们的关注 , 谷歌旗下人工智能公司 DeepMind 在研究《Highly accurate protein structure prediction with AlphaFold》中宣布 , 人们首次发现了一种通过计算来预测蛋白质结构的方法 。 即使在不知道相似结构的情况下 , AI 也可以在原子层面上精确预测蛋白质结构 。 也就是说 , 之前备受关注的 AlphaFold2 终于开源了 。
文章图片
无独有偶 , 作为相当热门的研究领域 , Science 同样在今天发表了一篇论文 , 介绍并开源了一个可媲美 AlphaFold2 的新工具 RoseTTAFold 。
所以说 , 赛马了 , 感兴趣的同学可以自由选择 。
AlphaFold2 开源 , 原子精度预测蛋白质结构
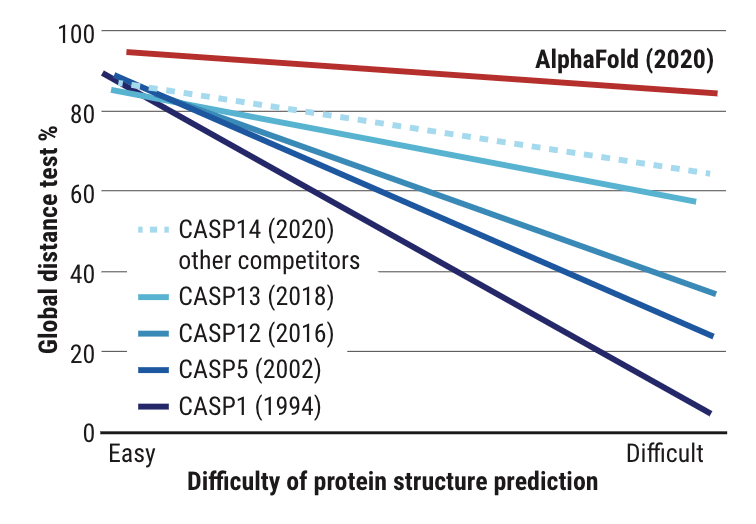
2020 年 12 月的国际蛋白质结构预测竞赛 CASP, 一项重磅成果引发了科技界所有人的关注:由 DeepMind 开发的 AlphaFold 2 击败一众选手 , 在准确性方面达到比肩人类实验结果 , 被认为是蛋白质折叠问题的解决方案 。
文章图片
在两年一次的 CASP 竞赛中 , 各组争先预测蛋白质的 3D 结构 。 2020 , AlphaFold 击败了所有其他小组 , 并在准确性方面与实验结果相匹配 。 它能以就计算机方法而言前所未有的准确度根据蛋白质的氨基酸序列预测其三维结构 。
这破解了出现五十年之久的蛋白质分子折叠问题 , 同时证明了 AI 对于科学发现 , 尤其是基础科学研究的影响 。
科学家们纷纷表示 , 这项突破极具意义 。 Alphafold 的突破性研究成果将帮助科研人员弄清引发某些疾病的机制 , 并为设计药物、农作物增产 , 以及可降解塑料的「超级酶」研发铺平道路 。
因此 , 这段时间以来 , 科研圈也在等待 AlphaFold 2 的技术细节 。
不久之前 , Demis Hassabis 就曾在 Twitter 上表示 DeepMind 将开源 AlphaFold2 , 如今终于兑现承诺 。
7 月 15 日 , Demis Hassabis、John Jumper 等人在 Nature 杂志上发表了文章《Highly accurate protein structure prediction with AlphaFold》 , 描述并开源了 AlphaFold2 , 它预测的蛋白质结构能达到原子水平的准确度 。

文章图片
- 链接:https://www.nature.com/articles/s41586-021-03819-2
- 开源地址:https://github.com/deepmind/alphafold
作为通讯作者之一 , Demis Hassabis 在一段声明中写到 , 「去年在 CASP14 大会上我们揭晓了一个可以将蛋白质 3D 结构预测精确到原子水平的全新 AlphaFold 系统 , 此后我们承诺会分享我们的方法 , 并为科学共同体提供广泛、免费的获取途径 。 今天我们迈出了承诺的第一步 , 在《自然》期刊上分享 AlphaFold 的开源代码 , 并发表了系统的完整方法论 , 详尽细致说明 AlphaFold 是如何做到精确预测蛋白质 3D 结构的 。 作为一家致力于推动科学进步的公司 , 我们期待看到我们的方法将为科学界启发出什么其他新的研究方法 , 也期待很快能和大家分享更多我们的新进展 。 」
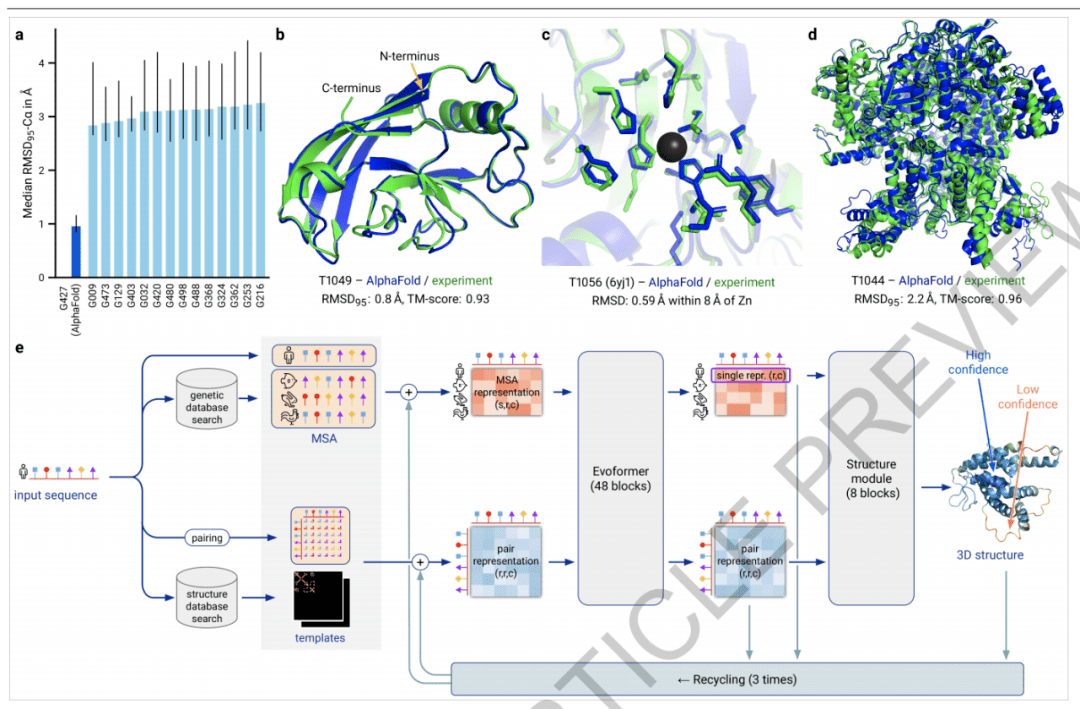
AlphaFlod 首次参加 CASP 就在 98 名参赛者中名列榜首 , 准确地从 43 种蛋白质中预测出了 25 种蛋白质的结构 。 而同组比赛中获得第二名的参赛者仅准确预测出了 3 种 。 AlphaFold 专注于从头开始建模目标形状 , 且并不使用先前已经解析的蛋白质作为模板 。 在大多数情况下 , AlphaFold 的准确性与实验相媲美 , 大大优于其他方法 。
图 1:AlphaFold 产出高准确度的架构 。
文章图片
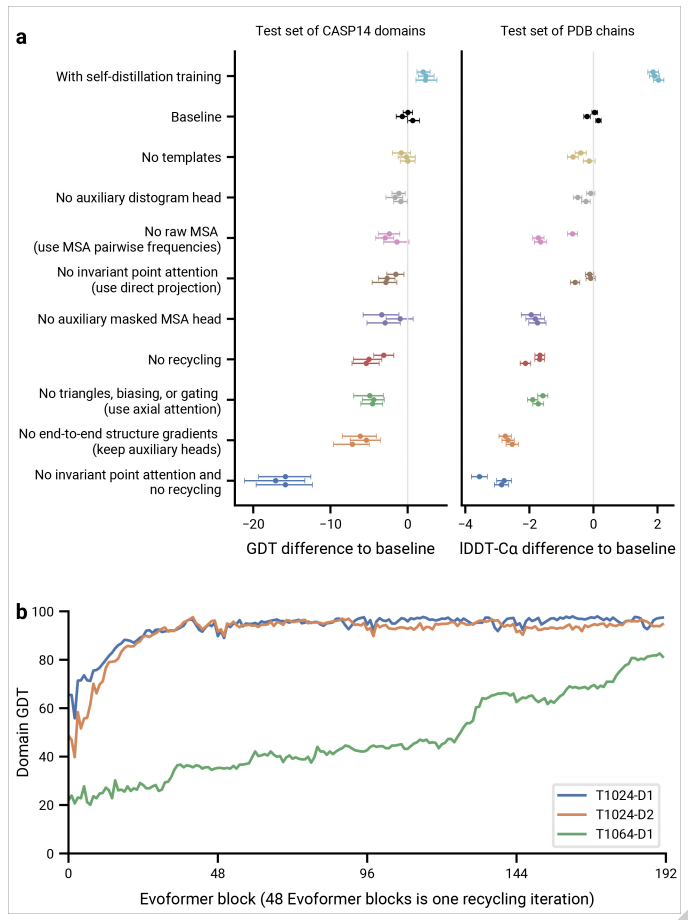
AlphaFold 网络直接预测给定蛋白质的所有重原子的三维坐标 , 使用基本氨基酸序列和同源序列的对齐序列作为输入 (如图 1e) 。
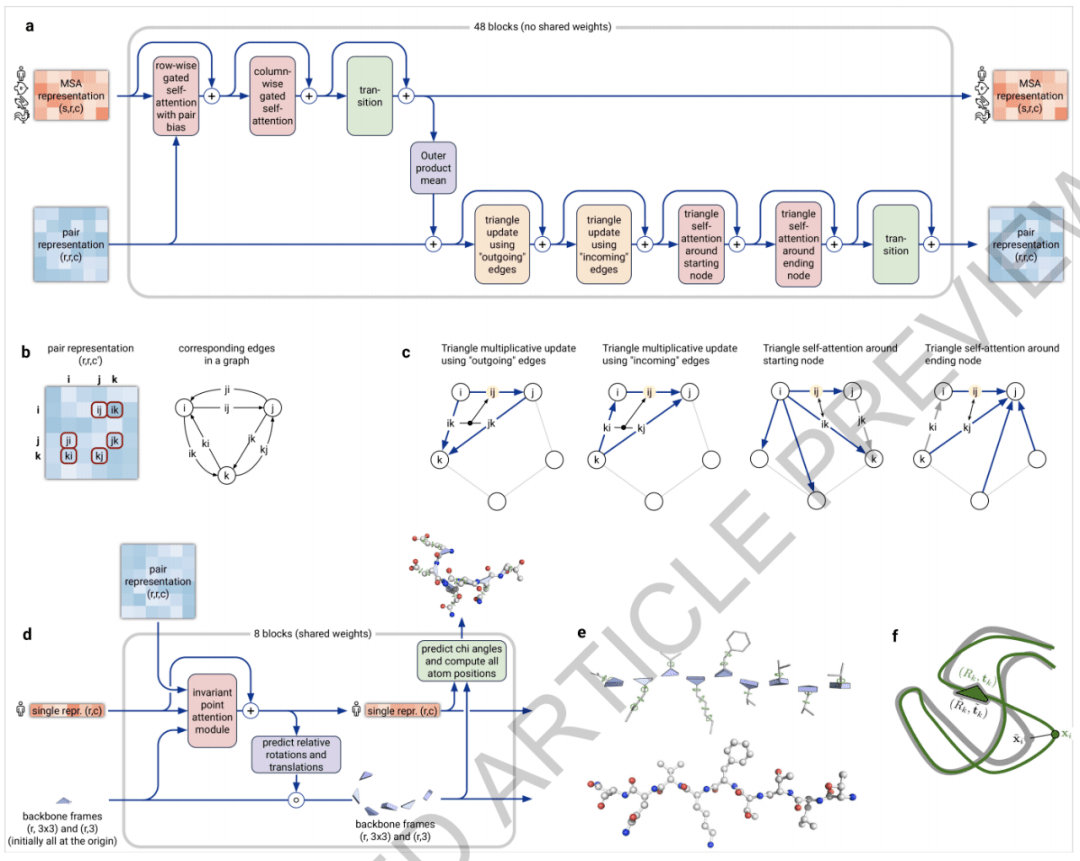
AlphaFold 网络由两个主要部分组成 。 首先 , 网络的主干通过一个称为 Evoformer 的新神经网络块的重复层来处理输入 , 产生一个 Nseq × Nres 阵列 (Nseq: 序列数 , Nres: 残差数), 它表示一个处理过的 MSA 和一个表示剩余对的 Nres × Nres 阵列 。 Evoformer 块包含许多新颖的基于注意力和非基于注意力的成分 , 它的关键创新是与 MSA 交换信息的新机制 , 并能直接推理空间和进化关系的配对表征 。
网络的主干之后是结构模块(Structure Module) , 该模块以蛋白质的每个残基的旋转和平移的形式引入了显式的 3-D 结构 。 这些表征在微不足道的状态下初始化 , 所有旋转设置为同一性(identity) , 所有位置设置为原点 , 但能够快速开发和完善具有精确原子细节的高度准确的蛋白质结构 。 这部分网络的关键创新包括打破链原子结构 , 允许同时局部细化结构的所有部分 , 一个新的「equivariant transformer」允许网络隐式地推理未表示的侧链原子 , 以及损失项可对残基方向的正确性赋予重要权重 。
图 3:架构细节
文章图片
图 4:解释神经网络
文章图片
更多细节大家可以查看 Nature 原文与 DeepMind 提供的补充材料 。
最后提一句 , 也不知是何原因 , 该论文未经编辑就出版了(5 月接收、7 月发表) , 难道是知道今天 Science 也将发表论文介绍一个可与 AlphaFold2 相匹配的研究?也就是下面这一篇 。
华盛顿大学等开发媲美 AlphaFold2 的新工具 RoseTTAFold
DeepMind 在 2020 年的 CASP14 会议上展示了其在该领域的显著成果 AlphaFold2 , 当时该技术在预测蛋白质方面取得了排名第一的准确率 。
华盛顿大学医学院蛋白质设计研究所(Institute for Protein Design)的研究者们很大程度上重现了 DeepMind 在蛋白质预测任务上的表现 , 他们联合哈佛大学、德克萨斯大学西南医学中心、剑桥大学、劳伦斯伯克利国家实验室等机构研发出了一款基于深度学习的蛋白质预测新工具 RoseTTAFold , 在预测蛋白质结构上取得了媲美 AlphaFold2 的超高准确率 , 而且速度更快、所需要的计算机处理能力也较低 。
这项研究已经在 Science 上发表 。

文章图片
论文链接:https://science.sciencemag.org/content/early/2021/07/14/science.abj8754
华盛顿大学医学院团队也向社区开源了该工具 , 来自世界各地的科学家都可以使用它来构建蛋白质模型 , 加速自己的研究 。 在上传至 GitHub 后不久 , 该工具就已被 140 多个独立研究团队下载 。
文章图片
项目地址 https://github.com/RosettaCommons/RoseTTAFold
具体地 , 在这项研究中 , 华盛顿大学医学院生物化学系教授、蛋白质设计研究所所长 David Baker 领导的计算生物学家团队开发了一款叫做「RoseTTAFold」的软件工具 , 该工具利用深度学习技术 , 根据有限信息准确、快速地预测蛋白质结构 , 原本这一工作需要数年的实验室研究 。
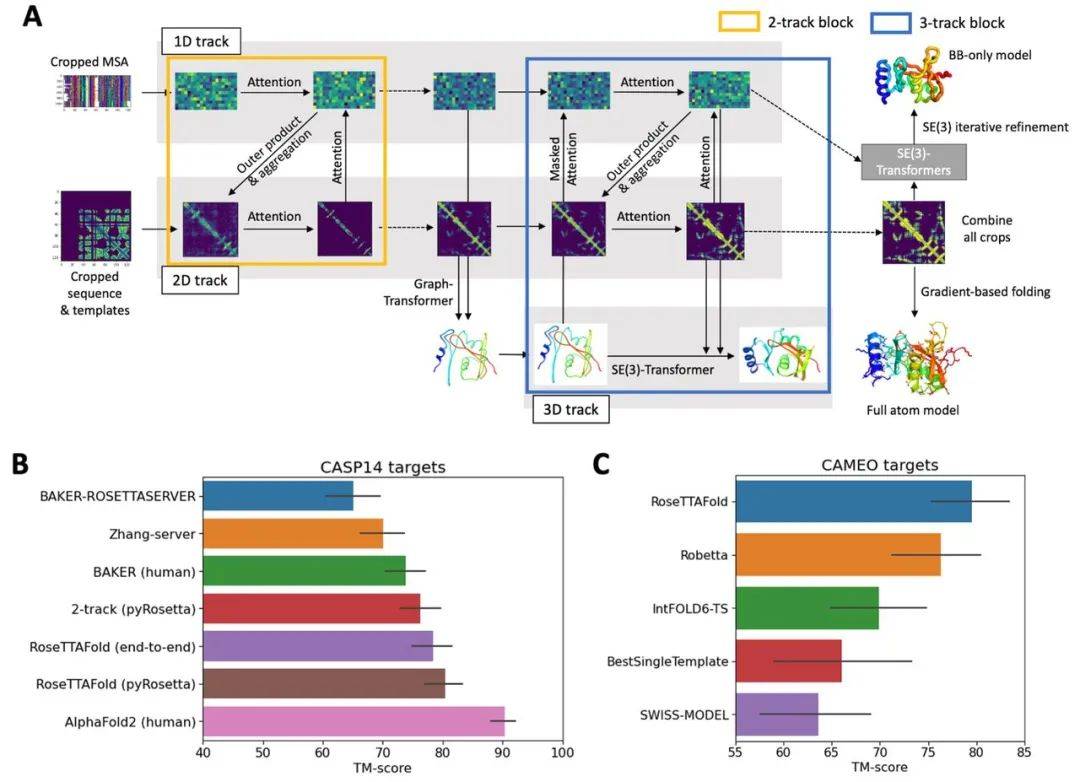
从结构上来看 , RoseTTAFold 是一个三轨(three-track)神经网络 , 意味着它可以兼顾蛋白质序列的模式、氨基酸如何相互作用以及蛋白质可能的三维结构 。 在这种结构中 , 一维、二维、三维信息来回流动 , 使得网络能够集中推理蛋白质的化学部分与它的折叠结构 。
下图 A 为具有 1D、2D 和 3D 注意力轨迹(attention track)的 RoseTTAFold 架构;B 为 CASP14 目标上蛋白质预测方法的平均 TM-score;C 为 CAMEO 中介(medium)和硬(hard)目标的盲基准测试结果 。
文章图片
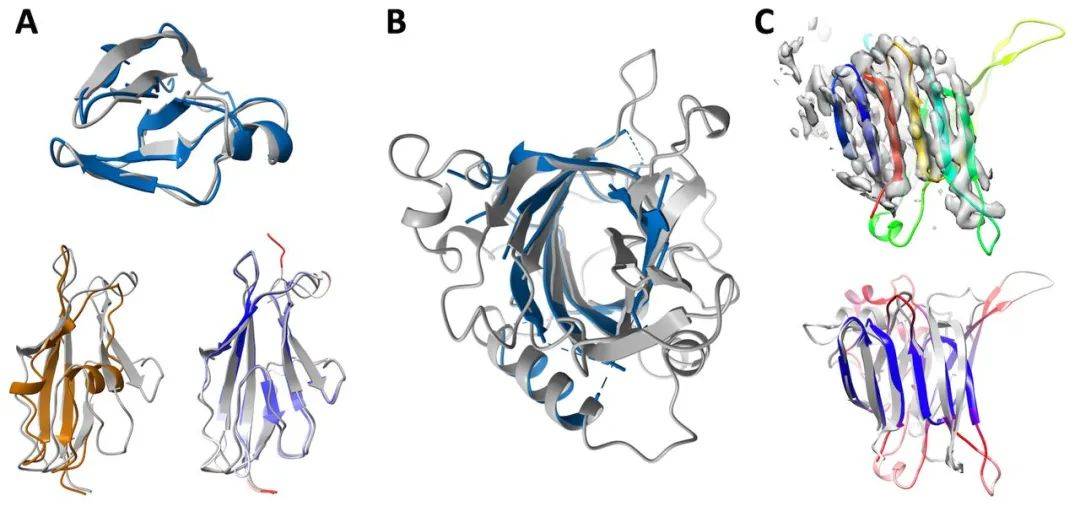
RoseTTAFold 方法的准确率比目前可用的方法高得多 , 因而研究者想要测试是否可以利用它解决以前未解决且具有挑战性的 MR 问题 , 并改进临界个案的解决方案 。 四个最近的晶体数据集 , 包括牛属甘氨酸 N - 酰基转移酶(GLYAT)、细菌氧化还原酶以及细菌表面层蛋白(SLP)(下图 A)和来自真菌平革菌属金孢子菌属的分泌蛋白(下图 B) , 基于 PDB 蛋白质数据库中可用的模型无法利用 MR 解决 , 因此研究者使用 RoseTTAFold 模型进行了重新分析:
文章图片
另一方面 , RoseTTAFold 能够利用一台游戏计算机在短短 10 分钟内计算出蛋白质结构 。 研究者使用 RoseTTAFold 计算出了数百种新的蛋白质结构 , 其中包括许多人类基因组中认知甚少的蛋白质 。 此外 , 他们还生成了与人类健康直接相关的一些蛋白质结构 , 包括与有问题的脂质代谢、炎症和癌细胞生长相关的蛋白质 。 他们还表明 , RoseTTAFold 可以用于建立复杂生物的模型 , 所需时间只是以前所需时间的一小部分 。
下图为使用 RoseTTAFold 的蛋白质预测流程 。 其中 , A 和 B 是从序列信息中预测大肠杆菌蛋白复合物的结构;C 表示由 RoseTTAFold 生成的 IL-12R/IL-12 复合结构符合以前发表的低温电子显微镜(cryo-EM)密度(EMD-21645) 。
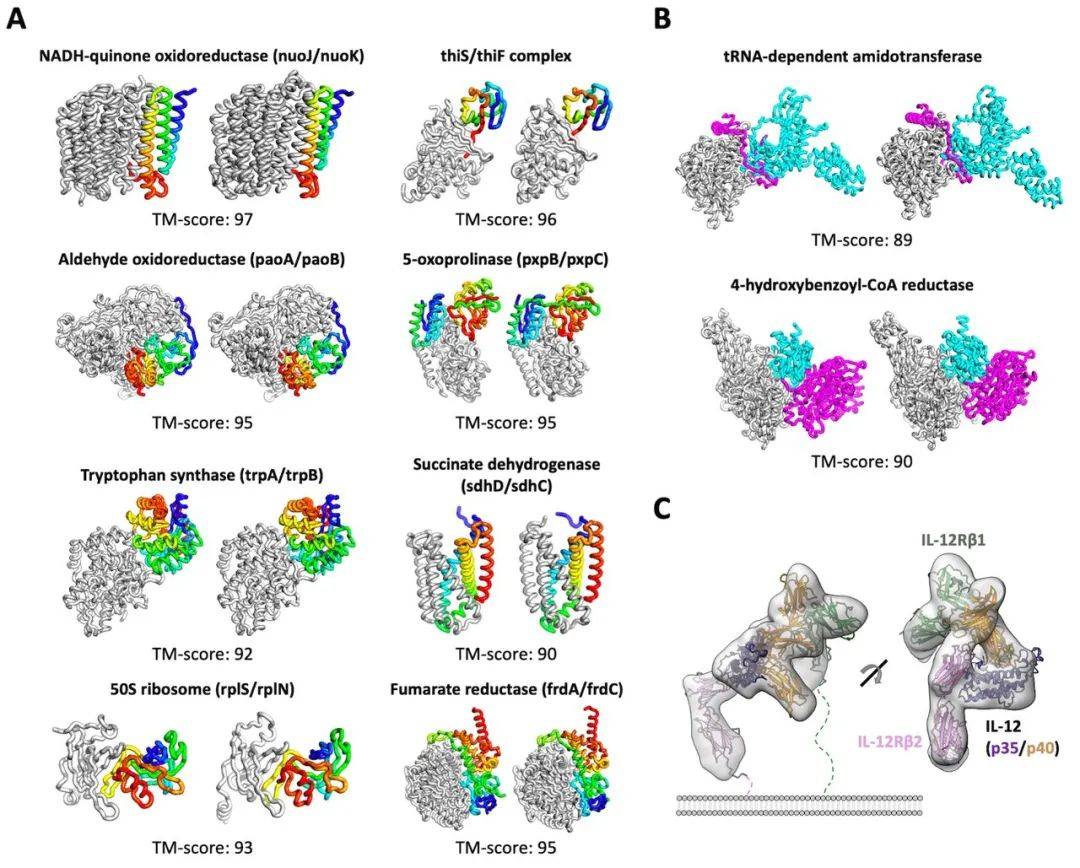
文章图片
论文一作、华盛顿大学博士后研究员 Minkyung Baek 表示:「我们希望这个新工具将造福整个研究领域 。 」
参考链接:
https://newsroom.uw.edu/news/accurate-protein-structure-prediction-now-accessible-all
【蛋白|一天之内,两大AI预测蛋白结构算法开源,分别登上Nature、Science】https://www.nature.com/articles/s41586-021-03819-2
推荐阅读







- 观测|期待!新年第一天火星合月,1月4日一起去看流星雨
- 水星|期待!新年第一天火星合月,1月4日一起去看流星雨
- 蛋白|二代新冠疫苗来了!打了一代,还需要它吗?
- IT|研究:高速公路延误可在一天内造成高达2.5亿美元的经济损失
- 大豆|烟台双塔食品股份有限公司 低钠高钙零大豆过敏原豌豆分离蛋白研究及产业化通过科技成果鉴定的公告
- IT|阿联酋卫生和预防部批准紧急使用国药第二代重组蛋白新冠疫苗
- IT|地图数据采集员 会开车就能赚钱的兼职:一天收入七八百
- 蛋白|血浆蛋白+氧化石墨烯能打印人造肌肉
- 最新消息|Movano智能戒指发布 希望有一天能监测慢性疾病
- 团队|从早到晚,看蓝月亮“除菌护卫队”如何呵护你的一天!